Fluoreszenz ist die Eigenschaft von Atomen und Molekülen, sogenannten Fluorophoren, Licht bei einer bestimmten Wellenlänge zu absorbieren und anschließend Licht mit längerer Wellenlänge zu emittieren. Fluoreszenzmikroskopie kann entweder auf Autofluoreszenz oder der Zugabe von Fluoreszenzfarbstoffen basieren. Sie wird hauptsächlich in der Biologie und Medizin eingesetzt, um Strukturen und Prozesse im Inneren einer Probe zu beobachten.
Einführung
1852 beobachtete der irische Physiker und Mathematiker Sir George Gabriel Stokes erstmals Fluoreszenz, als Sonnenlicht, das durch ein violettes Glasfenster gefiltert wurde, auf eine Flasche mit Chininwasser traf und blaues Licht emittiert wurde. Stokes bemerkte dieses blaue Licht, weil es ein Glas Weißwein durchquerte, welches das violette Licht aus dem Fenster herausfilterte und nur das blaue Licht des Chinins zurückließ.1 Stokes' Beobachtung veranschaulicht auch das Prinzip des Fluoreszenzmikroskops – ohne die Lichtfilterung durch das lila Glasfenster und das Weißweinglas hätte Stokes keine Fluoreszenz wahrnehmen können. Am Beispiel von Stokes' Beobachtung und dem grün fluoreszierenden Protein (GFP) wird in diesem Artikel die Fluoreszenz- und Fluoreszenzmikroskopie erläutert.
Das Prinzip der Fluoreszenz
Ein Fluorophor ist ein Molekül mit der Fähigkeit zur Fluoreszenz. Das bedeutet, dass das Molekül Photonen oder Lichtteilchen unterschiedlicher Wellenlängen absorbieren und emittieren kann. So konnte beispielsweise das Chinin in Stokes' Glas das violette Licht absorbieren und blaues Licht abgeben. Fluoreszenz kann daher definiert werden als die Emission von Licht (Photonen) mit einer Wellenlänge, die sich aus der Absorption von Licht (Photonen) mit einer anderen, typischerweise kürzeren Wellenlänge ergibt. Auf atomarer Ebene bewirkt die Absorption eines Photons durch ein Elektron im Fluorophor, dass das Elektron in eine Umlaufbahn springt, die weiter vom Atomkern entfernt ist (d. h. in einen energetisch höheren bzw. angeregten Zustand) (Abb. 1).
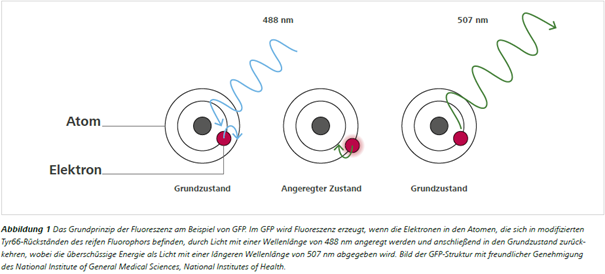
Im angeregten Zustand ist ein Elektron jedoch instabil, und wenn es in seinen Grundzustand zurückkehrt, wird ein Photon mit längerer Wellenlänge emittiert, um die überschüssige Energie abzubauen – dies wird als Fluoreszenz bezeichnet.
Die Zustände und Übergänge von Elektronen in einem Fluorophor sind komplexer als oben beschrieben, lassen sich aber einfach mit dem Jablonski-Diagramm veranschaulichen, das 19332 vom polnischen Physik-Professor Aleksander Jablonski entworfen wurde. Im Jablonski-Diagramm (Abb. 2, oben) zeigt ein blauer vertikaler Pfeil die Absorption eines Photons durch ein Molekül, wodurch ein Elektron innerhalb von Femtosekunden (10 –15 s) vom Grundzustand (S0) in einen Singulett-Zustand mit höherer Energie (S1und S2) springt. Molekulare Schwingungen, bei denen die internuklearen Abstände im Laufe der Zeit variieren, bewirken weiterhin, dass das Elektron in jedem Singulett-Zustand in diskreten Energieniveaus existiert. Diese werden als Schwingungsebenen bezeichnet. Als Ergebnis dieser diskreten Energiezustände können Elektronen Photonen mit einem Wellenlängenbereich absorbieren, die das Elektron in ein höheres Energieniveau verschieben können. Der Wellenlängenbereich, der auf diese Weise Fluoreszenz induzieren kann, steht in direktem Zusammenhang mit dem Anregungsspektrum eines Fluorophors (Abb. 2, unten).
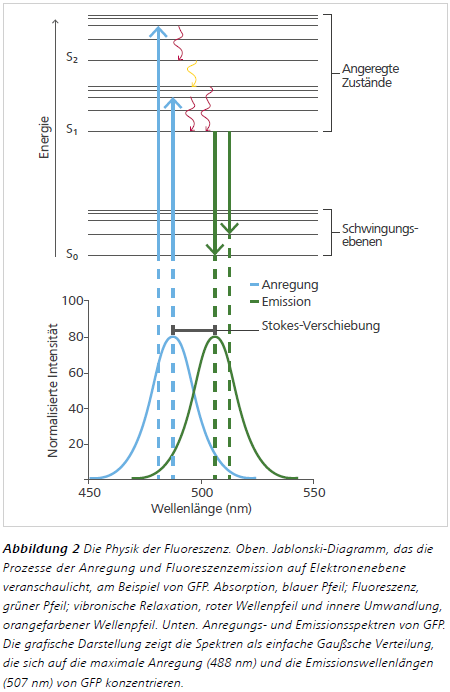
Nach der Anregung gibt es mehrere Möglichkeiten, wie ein Elektron die überschüssige Energie ableiten und in seinen Grundzustand zurückkehren kann. Zwei solcher Wege, im Jablonski-Diagramm durch geschwungene Pfeile gekennzeichnet, sind die innere Umwandlung und die vibronische Relaxation (Abb. 2, oben). Dies sind strahlungslose Energieverluste, die innerhalb von Pikosekunden (10-12 s) auftreten. Bei der vibronischen Relaxation kehrt das Elektron auf das niedrigste Energieniveau seines aktuellen Singulett-Zustandes zurück, indem es Schwingungsenergie auf benachbarte Moleküle überträgt, während die innere Umwandlung den Übergang zwischen Singulett-Zuständen (z. B. S2 zu S1) darstellt. In effizienten Fluorophoren führt die Energiedissipation zwischen S1 und dem Grundzustand zur Emission eines Photons und erfolgt innerhalb von Nanosekunden (10-9 s). Da ein Elektron zu jedem beliebigen Schwingungsniveau des Grundzustandes zurückkehren kann, können Photonen mit unterschiedlichen Wellenlängen emittiert werden (bekannt als das „Emissionsspektrum“ des Fluorophors, Abb. 2, unten).
Und schließlich hat das emittierte Photon eine geringere Energie (längere Wellenlänge) als das absorbierte Photon, da ein Teil der Anregungsenergie des Elektrons durch strahlungslose Prozesse abgebaut wird. Dies wird als „Stokes-Verschiebung“ bezeichnet (Abb. 2, unten) und entspricht dem, was Stokes beobachtete, als Chinin durch violettes Licht angeregt wurde und blau emittierte.
Fluorophore und Immunfluoreszenz
Im Jahr 1962 wurde entdeckt, dass die fluoreszierende Eigenschaft der Quallenart Aequorea victoria (Abb. 3) auf das grün fluoreszierende Protein (GFP) zurückzuführen ist.3
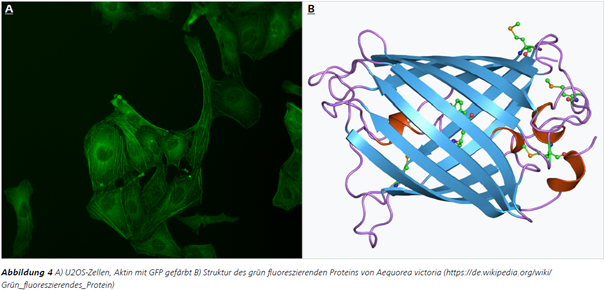
GFP ist ein kleines 27-kDa-Protein, das zu einer β-Barrel-Struktur gefaltet ist (Abb. 4).
Der reife Fluorophor ragt in den Hohlraum dieses β-Barrels hinein. Nach der Translation des Proteins bildet sich der Fluorophor spontan, indem sich die Struktur eines Tripeptides (mit Tyr66 in der Mitte) verändert. Seit den 1990er-Jahren wurde die Aminosäuresequenz und -struktur des Wildtyp-GFP fortlaufend mutiert, um fluoreszierende Proteine mit höherer Stabilität und Helligkeit sowie einer Reihe verschiedener Farben zu erzeugen.4 Fluoreszierende Proteine können in einer Vielzahl von Anwendungen entweder als Reporter- oder Fusionsproteine eingesetzt werden und dienen der Analyse der Proteinlokalisation sowie der Untersuchung einer Reihe zellulärer Prozesse innerhalb lebender Zellen.
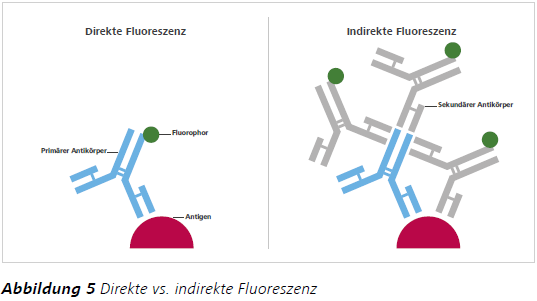
Vor der Entdeckung von GFP und auch heute noch werden kleine organische Moleküle als Fluoreszenzmarker verwendet. Diese basieren meist auf der Struktur von Xanthen- und Cyaninfarbstoffen, aber es existiert eine Vielzahl solcher Marker. Einige sind empfindlich gegenüber ihrer Umgebung, was sie zu attraktiven Sensoren für pH-Wert, Spannung oder Ionenkonzentration macht. Andere kleine organische Farbstoffe werden als Marker für Organellen (z. B. Mito- und LysoTracker-Farbstoffe) oder die DNA (z. B. die fluoreszierenden DNAInterkalatoren Ethidiumbromid, 4,6-Diamidin-2-phenylindol (DAPI) und Hoechst) verwendet. Im Jahr 1941 gelang ein wichtiger Durchbruch in der Verwendung kleiner organischer Farbstoffe als Fluoreszenzmarker, als diese erstmals an Antikörper konjugiert wurden und somit das Feld der Immunfluoreszenz einleiteten.5 Die Immunfluoreszenz ist eine der am weitesten verbreiteten biologischen Techniken der Fluoreszenzmikroskopie. Um ein Antigen von Interesse zu visualisieren, werden Zellen oder Gewebe mit einem an ein Fluorophor konjugierten Antikörper inkubiert. Der Fluorophor kann entweder an den primären Antikörper, der das Antigen erkennt, oder an einen sekundären Antikörper, der den primären Antikörper erkennt, konjugiert werden. Dies wird als direkte bzw. indirekte Immunfluoreszenz bezeichnet (Abb. 5). Vorteile der direkten Immunfluoreszenz sind ein kürzeres Protokoll und eine geringere Gefahr der Kreuzreaktivität; dergrößte Nachteil ist die geringe Signalintensität. Bei der indirekten Immunfluoreszenz können mehrere sekundäre Antikörper den gleichen primären Antikörper binden, um das Fluoreszenzsignal zu verstärken. Dieses zweistufige Labeling-Protokoll erhöht jedoch auch die Komplexität und kann zu Kreuzreaktionen innerhalb der Probe führen.
Das Fluoreszenzmikroskop
Die Hauptaufgabe eines Fluoreszenzmikroskops besteht darin, eine Probe mit Licht einer Anregungswellenlänge zu beleuchten und gleichzeitig das vergleichsweise schwächere emittierte Licht der Probe zu sammeln und zu trennen. Im Beispiel von Stokes' Beobachtung werden diese Aufgaben durch das violett gefärbte Glasfenster bzw. das Glas Weißwein erfüllt. Im Fluoreszenzmikroskop sorgt der Filterwürfel für diese Trennung des Lichts.Abbildung 6 veranschaulicht den grundlegenden Aufbau eines inversen Weitfeld-Fluoreszenzmikroskops, das eine Probe mit GFP abbildet.
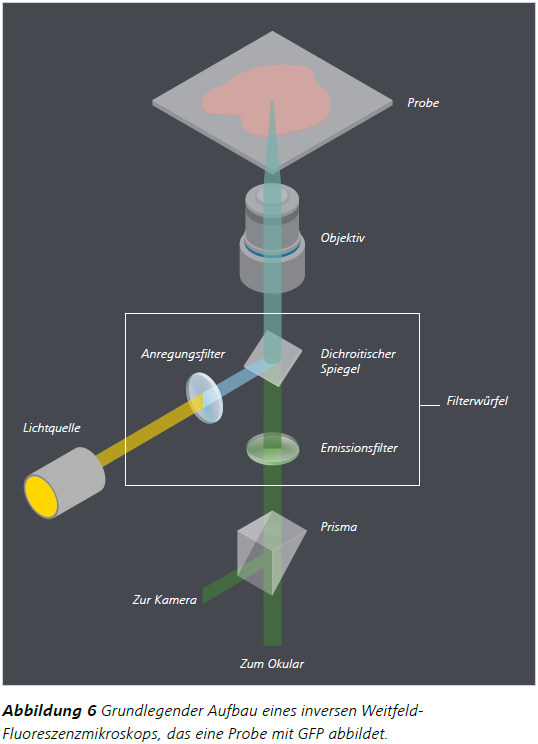
Die erste wichtige Komponente ist die Lichtquelle. Um die optimale Wellenlänge des Anregungslichts für einen bestimmten Fluorophor zu erzeugen, wird ein Anregungsfilter in den Lichtweg zwischen Lichtquelle und Probe eingesetzt. Dies ist die erste von drei Komponenten im Filterwürfel, der aus einem Anregungsfilter, einem dichroitischen Teilerspiegel und einem Emissions- oder Sperrfilter besteht. Der Anregungsfilter ist in der Regel ein Bandfilter, d. h., er ist durchlässig für Licht mit einem engen Wellenlängenbereich und blockiert andere Wellenlängenbereiche.
Nach der Filterung wird das Anregungslicht von einem dichroitischen Teilerspiegel auf die Probe reflektiert. Dabei handelt es sich um einen weiteren Filter, der in einem 45°-Winkel sowohl zur Lichtquelle als auch zur Probe steht. Die spektralen Eigenschaften des dichroitischen Teilerspiegels sind so, dass das Anregungslicht auf die Probe reflektiert und das emittierte Licht mit längerer Wellenlänge durch den Filter zum Detektor übertragen wird. Daher muss der dichroitische Teilerspiegel für eine optimale Bildgebung auch den Anregungsund Emissionsspektren (d. h. die Stokes-Verschiebung) des gewählten Fluorophors entsprechen.
Auf dem Weg zur Probe wird das Anregungslicht im Objektiv fokussiert. Im Weitfeld-Fluoreszenzmikroskop dient das Objektiv sowohl als Kondensor als auch zur Vergrößerung der Probe. Darüber hinaus fängt das Objektiv das von der Probe emittierte Licht ein und sendet es zum dichroitischen Teilerspiegel im Filterwürfel zurück.
Obwohl bereits der dichroitische Teilerspiegel verhindern soll, dass Anregungslicht den Detektor erreicht, wird oft zusätzlich ein Emissionsfilter zwischen diesen Komponenten eingesetzt, um externes Anregungslicht oder Hintergrundfluoreszenz zu blockieren. Bei der Abbildung eines einzelnen Fluorophors kann es sich bei dem Emissionsfilter um einen Langpassfilter handeln, der Licht mit längeren Wellenlängen durchlässt und gleichzeitig kürzere Wellenlängen blockiert. Allerdings sind sowohl die Anregungs- als auch die Emissionsfilter in der Regel Bandpassfilter mit einem engen Bereich transmittierter Wellenlängen. Dies ermöglicht die Abbildung von mehr als einem Fluorophor in einer Probe.
Der Aufbau des Filterwürfels wird komplexer, wenn mehr als ein Fluorophor abgebildet werden soll. Das Fluoreszenzmikroskop kann mehrere Filterwürfel aufnehmen, die auf die Anregungs- und Emissionsspektren einer Reihe von Fluorophoren abgestimmt sind. Der Wechsel zwischen den Filterwürfeln ermöglicht die Abbildung mehrerer Fluorophore in einer Probe. Schon ein geringer Unterschied in der Ausrichtung der Filterwürfel kann jedoch zu einer Fehlausrichtung der Bilder der verschiedenen Fluoreszenzkanäle führen. Um dieses Problem zu lösen, können die Anregungsfilter auf Filterrädern montiert werden, die für alle Fluoreszenzkanäle den gleichen dichroitischen Spiegel verwenden. Dies löst das Ausrichtungsproblem und ermöglicht ein schnelleres Umschalten zwischen Anregungs- und Emissionsfiltern. Es bedeutet aber auch, dass der dichroitische Teilerspiegel, in diesem Fall als polychromatischer Teilerspiegel bezeichnet, die Anregungswellenlängen für mehrere Fluorophore spezifisch reflektieren muss, während er gleichzeitig das Emissionssignal für jedes dieser Elemente sendet. Der Schlüssel zur Verwendung mehrerer Fluorophore in einem Experiment liegt darin, diejenigen auszuwählen, deren Emissionsspektren sich so wenig wie möglich überschneiden.
Die Fluoreszenzmikroskopie ist aufgrund ihres breiten Anwendungsspektrums und der relativ geringen Anforderungen seit Langem ein unverzichtbares Instrument zur Untersuchung aller Aspekte der Zell- und Molekularbiologie. Die Technik des Fluoreszenzmikroskops (Abb. 7) wird ständig weiterentwickelt und verbessert, um der Forschung noch zweckbestimmtere und genauere Mikroskope zur Verfügung stellen zu können.
Zusammen mit der kontinuierlichen Entwicklung optimierter und spezialisierter Fluoreszenzsonden macht dies die Fluoreszenzmikroskopie zu einem festen Bestandteil der biologischen Forschung.

Literaturverzeichnis
[1] Stokes, G. G. XXX. On the change of refrangibility of light. Philosophical Transactions of the Royal Society of London 142, 463 – 562 (1852).
[2] Jablonski, A. Efficiency of Anti-Stokes Fluorescence in Dyes. Nature 131, 839, doi:10.1038/131839b0 (1933).
[3] Shimomura, O., Johnson, F. H. & Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol 59, 223 – 239 (1962).
[4] Heim, R. & Tsien, R. Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol 6, 178 – 182 (1996).
[5] Coons, A. H., Creech, H. J. & Jones, R. N. Immunological properties of an antibody containing a fluorescent group. Proceedings of the Society for Experimental Biology and Medicine 47, 200 – 202 (1941).
Empfohlene Übersichtsarbeiten zur weiteren Lektüre
- Chudakov, D. M., et al.: Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev 90(3): 1103 – 1163 (2010). Umfangreiche Abhandlung der Struktur, Varianten, Eigenschaften und Anwendungen verschiedener fluoreszierender Proteine.
- Lichtman, J. W., und Conchello, J. A. Fluorescence microscopy. Nat Methods 2(12), 910-919 (2005). Hervorragender Kurzartikel über die Grundprinzipien der Fluoreszenz und des Fluoreszenzmikroskops.
- Sanderson, M. J., et al.: Fluorescence microscopy. Cold Spring Harb Protoc 2014(10): pdb top071795 (2014). Detailreiche Erläuterung der Prinzipien und Eigenschaften verschiedener Fluoreszenzmikroskop-Bildgebungen.
- Shaner, N. C., et al.: A guide to choosing fluorescent proteins. Nat Methods 2(12): 905 – 909 (2005). Kurze Besprechung mit hilfreichen Tabellen, um die für eine Anwendung passenden fluoreszierenden Proteine und deren Filtersets zu wählen.
- Waggoner, A.: Fluorescent labels for proteomics and genomics. Curr Opin Chem Biol 10(1): 62 – 66 (2006). Kurze Besprechung der Geschichte und der Charakteristiken von kleinen organischen Molekülen, die als Fluoreszenzmarker verwendet werden.
Empfohlene Links zur Weiterbildung
- http://zeiss-campus.magnet.fsu.edu/tutorials/matchingfiltersets/indexflash.html Interaktives Setup, in dem Wellenlängen und Lichtintensität angezeigt werden, die von verschiedenen Mikroskop-Lichtquellen emittiert werden. Es können auch Anregungsfilter Sets ausgewählt werden, die zu den Anforderungen des gewählten Fluorophors passen.
- http://zeiss-campus.magnet.fsu.edu/tutorials/filterwheel/indexflash.html Interaktive Illustration des Filterradprinzips.
- http://zeiss-campus.magnet.fsu.edu/articles/basics/fluorescence.html Hervorragende Beschreibung des Prinzips der Fluoreszenz, der Komponenten und der Eigenschaften eines Fluoreszenzmikroskops.
- http://zeiss-campus.magnet.fsu.edu/articles/probes/fpintroduction.html Umfangreiche Beschreibung der fluoreszierenden Proteine hinsichtlich ihrer Geschichte, Struktur, Adaptationen und Charakteristiken.